【文章速递】【Journal of Energy Chemistry】提高电池电解质设计中经典分子动力学模拟的可靠性
Posted at 2024-09-30 in 最新文章
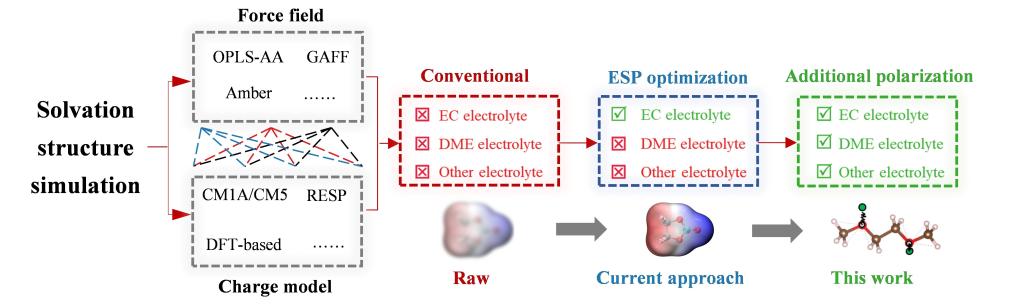
这项研究表明,使用 GAFF 和 OPLS-AA等常用力场进行经典分子动力学(CMD)模拟可能无法普遍有效地模拟所有类型的电解液,而这一问题往往没有引起关注。本研究以碳酸酯、醚和腈类电解液为例,发现 OPLS-AA力场由于不能很好地还原EC分子的静电势,导致其模拟的碳酸酯类电解液的溶剂化结构不准确。另一方面,尽管GAFF和OPLS-AA力场对于醚基和腈基溶剂分子的 静电势有很好的描述,但它们仍然难以准确模拟溶剂化结构。
我们的理论分析表明,这些力场缺乏极化效应描述是造成主流力场模拟溶剂化结构不准确的主要原因。模拟溶剂化结构的准确性不仅取决于静电势,还取决于如何描述极化。这一见解释了为什么在以往的工作中,研究人员优化静电势描述后也无法准确模拟溶剂化结构。
本研究证明,将简单的 Drude 极化模型与 GAFF 力场相结合,可以有效地模拟碳酸酯、醚和腈基电解质的溶剂化结构。与其他已有经验方法相比,这种方法具有更高的有效性和通用性。虽然一些现有的可极化力场也可提高 CMD 模拟的准确性,但复杂的可极化力场的速度通常要慢一个数量级,而且所支持软件的用户较少。因此,本工作希望提供新的改进思路,利用现有广泛使用的开源力场提高 CMD 模拟的可靠性。
发表时间:2024年9月
文章链接:
Improving the reliability of classical molecular dynamics simulations in battery electrolyte design
https://doi.org/10.1016/j.jechem.2024.09.038